How to Assess an Electronic Health Records (EHR) System for Clinical Research
Whether paper or eSource, investigators and sponsors need to ensure that there are adequate controls in place to preserve the confidentiality, integrity, and security of data.
Electronic Health Records (EHR) systems need to be assessed by the Sponsor and/or CRO for compliance with regulatory requirements before investigator sites activation, but how can this type of systems be assessed? What are the main challenges of EHR systems hosting source data for clinical studies?
This article reviews the latest Guidance for Industry published in Jul 2018 by the FDA “Use of Electronic Health Record Data in Clinical Investigations”1 and compares it with the Reflection paper released by the EMA on the same topic in 2010 “Expectations for electronic source data and data transcribed to electronic data collection tools in clinical trials”2.
The May 2007 FDA Guidance for Industry “Computerized Systems Used in Clinical Investigations” was instrumental in helping to ensure that records generated by a EHR system (understood as a computerized system used in clinical investigations to generate and maintain source data on each clinical trial subject) are GCP compliant and must meet the same fundamental elements of data quality (ALCOA+ Principles: Accurate, Legible, Contemporaneous, Original, Attributable, Complete, Consistent) that are expected of paper records3.
After reviewing in detail, the long-awaited Guidance for Industry on the “Use of Electronic Health Record Data in Clinical Investigations” released in July 2018, I do not believe it helps to clarify the multiple doubts around the use of Electronic Health Records (EHRs) as a source of data in clinical investigations.
The new Guidance states that the FDA encourages sponsors and clinical investigators to work with health care organizations to use the EHR and EDC systems that are interoperable or fully integrated and the Guidance content tends to revolve around interoperability1. Even though EHR-EDC systems integration it is recognized to be the future, it is not today’s most common environment.
What the new Guidance clarifies is that EHR systems are not under the control of FDA-regulated entities (e.g. sponsors, clinical investigators)1. The investigator sites had been dealing with the varying interpretations on how FDA’s Part 11 applied to EHRs, but FDA does not intend to assess compliance of EHRs systems with 21 CFR Part 11.
“21 CFR Part 11 is not mandatory for a site’s EHR system. It has been confused desirable characteristics of Part 11 as full Part 11 compliance”
The main point about EHRs systems is that they are developed and maintained by the health institutions for general patient medical records. Consequently, they are part of the practice of medicine which FDA does not regulate. However, since these EHR systems hold data relevant to clinical studies, they should have the major elements found in 21 CFR Part 11 that are pertinent to the integrity of data and this is what the new FDA Guidance highlights: “FDA’s acceptance of data from clinical investigations for decision-making purposes depends on FDA ability to verify the quality and integrity of the data during FDA inspections”1.
The EMA “Reflection paper on expectations for electronic source data transcribed to electronic data collection tools in clinical trials” published in 2010, in my opinion covers the topic following a more practical approach. The Reflection paper refers to the twelve requirements stated in the CDISC (Clinical Data Interchange Standards Consortium) standard for source data4, irrespective of the technology used to hold the data and provides recommendations on how to meet them2 (see Table 1).
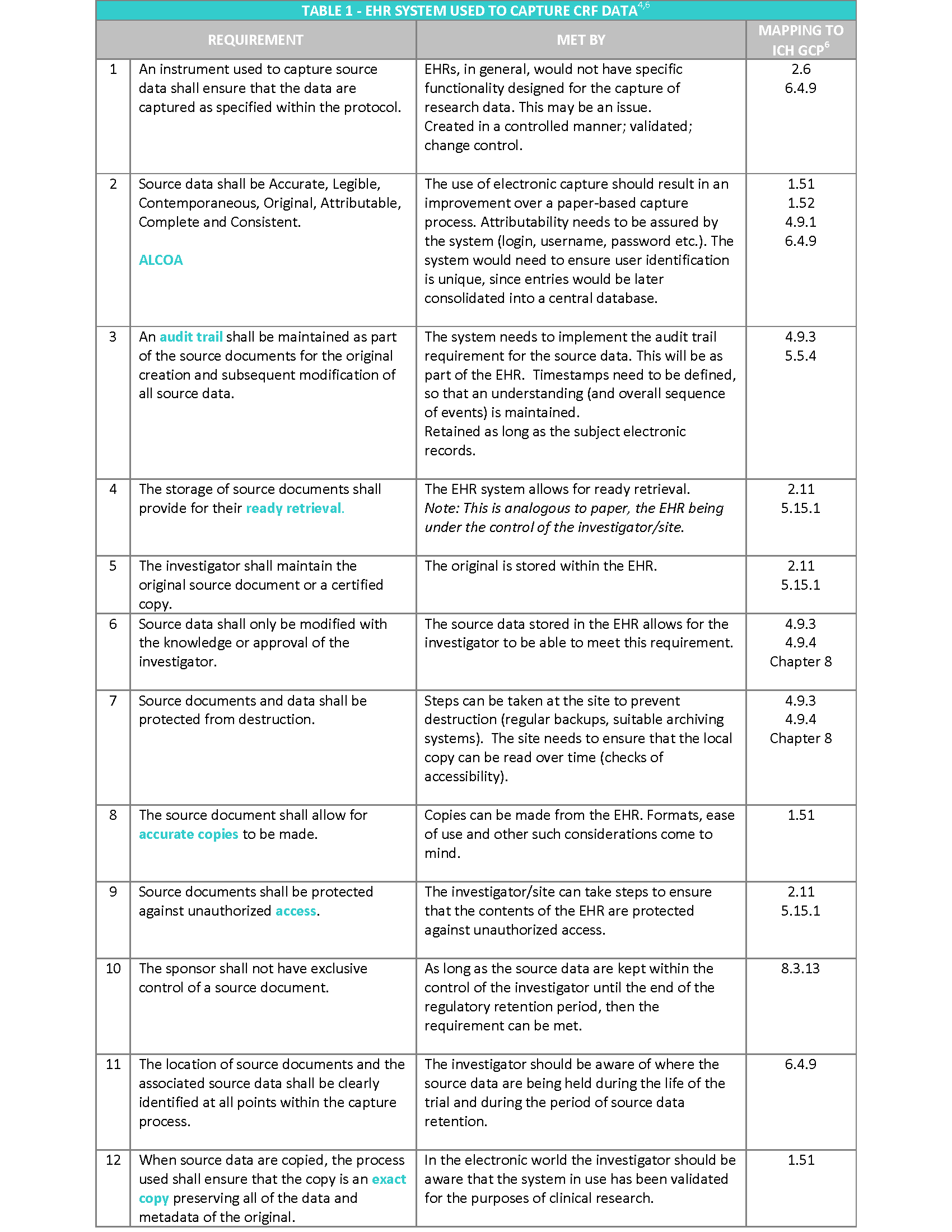
In addition, to the above requirements, the following aspects also need to be considered5:
- Access to the system should be available for inspectors and sponsor representatives (e.g. monitors and auditors), which is limited to trial patients. This will enable source data verification of clinical trial subjects whilst protecting the confidentiality of non-trial patients. This should include access to audit trails.
- Appropriate archiving to ensure long term reliability, retrieval and reproducibility of electronic data (and metadata), in line with regulatory storage timelines (Note: this will be 25 years as standard under the new EU Clinical Trials Regulation 536/2014).
Following the requirements discussed in this article and understanding the clinical trial source data requirements will help implementing an EHR systems that is robust and GCP compliant.
Lack of control over the source data could result in an inability to reconstruct the trial and the trial data being labelled as unreliable.
Conclusion
Even though we have regulatory guidance documents covering EHR systems hosting source data used in clinical investigations, there are still a lot of open questions on the expectations for EHR systems, how to assess them and how to mitigate the identified risks.
Whether you are an Investigator, CRA, Auditor, any other clinical research professional, if you want to gather a deeper knowledge on how to assess EHR systems to determine how well they meet the requirements of GCP and/or how to mitigate the risks for those systems that are not fully compliant, I would recommend reviewing the CDISC/ eSDI Group document “Leveraging the CDISC Standards to Facilitate the use of Electronic Source Data within Clinical Trials”4 in conjunction with the EMA Reflection paper2 , the FDA eSource Guidance for Industry7, and the other guidance documents referenced in this article.
References
1. FDA Guidance for Industry: Use of Electronic Health Record in Clinical Investigations, July 2018, US Food and Drug Administration (FDA).
2. EMA/INS/GCP/454280/2010 – Reflection paper on expectations for electronic source data and data transcribed to electronic data collection tools in clinical trials, 2010.
3. FDA Guidance for Industry: Computerized Systems Used in Clinical Investigations, May 2007, US Food and Drug Administration (FDA).
4. CDISC e-source standard requirements-CDISC (Clinical Data Interchange Standards Consortium). Version 1.0, 20 Nov 2006.
5. MHRA Position Statement and Guidance Electronic Health Records. Version 1.0. 6Sep2015
6. International Council for Harmonization (ICH): ICH Harmonized Guideline, Integrated Addendum to ICH E6(R1): Guideline for Good Clinical Practice E6(R2). Step 4, version 9 November 2016.
7. FDA Guidance for Industry: Electronic Source Data in Clinical Investigations, Sep 2013, US Food and Drug Administration (FDA).
Author: Dr Leire Zúñiga, Director and Principal GCP Consultant
PHARMITY, 26th September 2018
www.pharmity.com


